Introduction
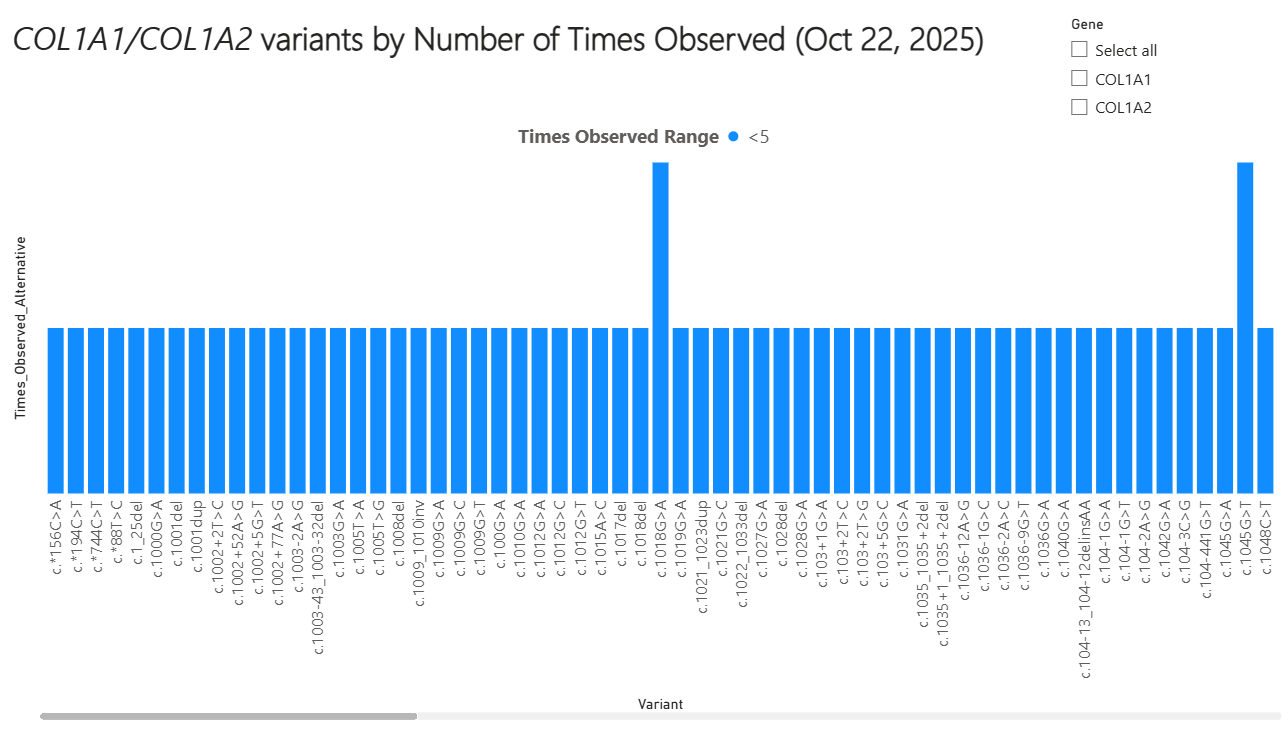
Osteogenesis imperfecta (OI) is a rare, hereditary skeletal dysplasia, with several subtypes, associated with bone fragility, growth deficiency, and variable secondary features [1]. The estimated diagnosed population prevalence of OI is between 5 to 7 per 100,000 individuals globally [2-7]. The majority of cases of OI (over 80%) are caused by pathogenic variants of the genes encoding type I collagen (COL1A1 and COL1A2) [8].
Skeletal manifestations of OI include lifelong predisposition to vertebral, long bone, and other fractures after relatively little to no trauma; skeletal deformities; short stature; and impaired mobility. Extraskeletal manifestations are common in tissues that have high levels of type I collagen expression [8-10].
Diagnosis of OI requires a multi-disciplinary approach; it is largely dependent on family history, the incidence of fractures, and other skeletal and extraskeletal signs (eg, blue sclerae, dentinogenesis imperfecta, and spine, long bone, and craniofacial deformities). Principal components of the diagnostic workup include the evaluation of family history, physical examination, and diagnostic imaging focusing on the skeletal system. Molecular confirmation of OI occurs through genetic testing [11-13].