Introduction
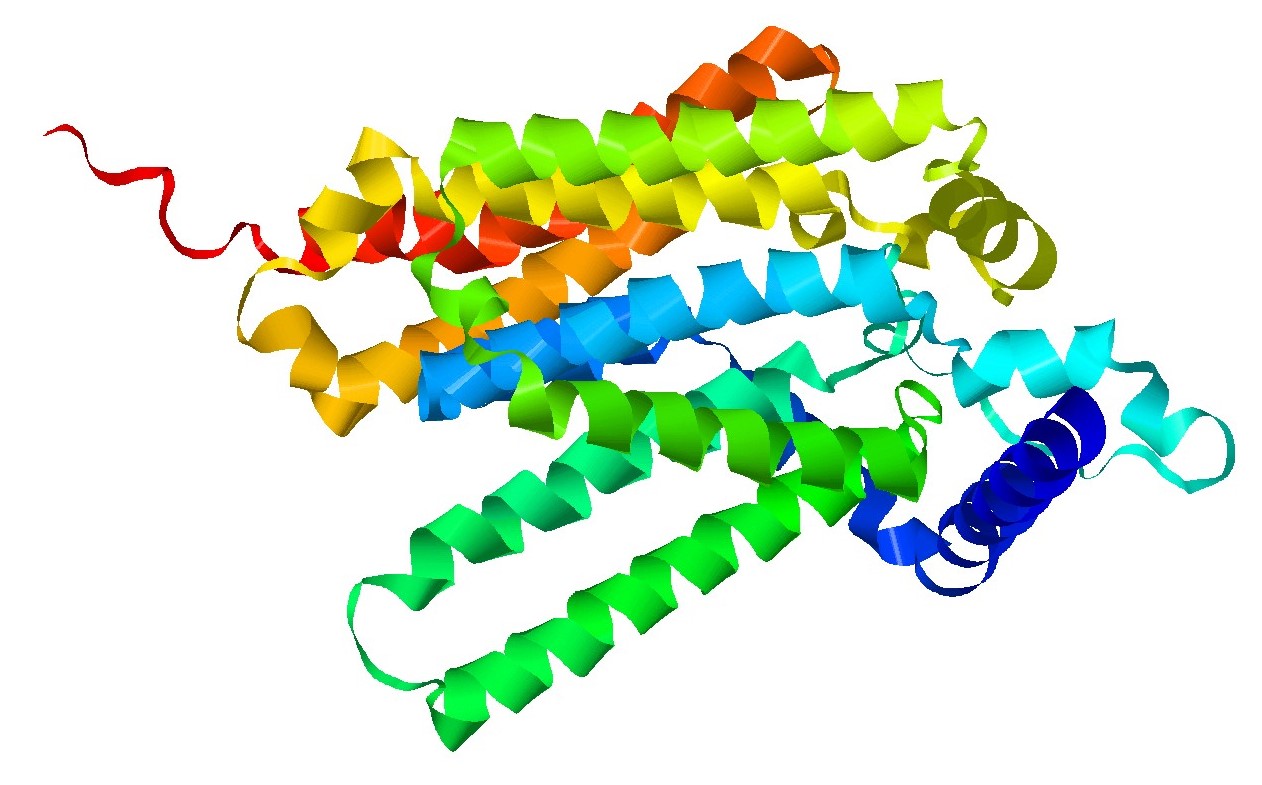
Glycogen storage disease type Ia (GSDIa, OMIM, #232200) also known as von Gierke disease, is a life-threatening, rare, inherited disorder of carbohydrate metabolism. The prevalence of all GSD type I is approximately 1 in 100,000 with GSDIa representing 80% of GSD type I. (1,2,3). GSDIa results from biallelic pathogenic variants in the G6PC1 gene (previously known as G6PC) (1, 4). Patients with GSDIa are deficient in glucose-6-phosphatase-α (G6Pase-α) catalytic activity, an enzyme required for the conversion of glycogen into glucose.
Patients with GSDIa experience symptoms such as severe ketotic hypoglycemia, lactic acidosis, hyperlipidemia, and hyperuricemia due to fasting intolerance (1,3). GSDIa is diagnosed based on clinical presentation, characteristic biochemical and laboratory findings, and the presence of two disease-associated variants in the G6PC1 gene (5). A liver biopsy deficient for G6Pase-α enzymatic activity also confirms the diagnosis of GSDIa and is less frequently used given the availability of non-invasive molecular genetic testing.