Home »
Introduction
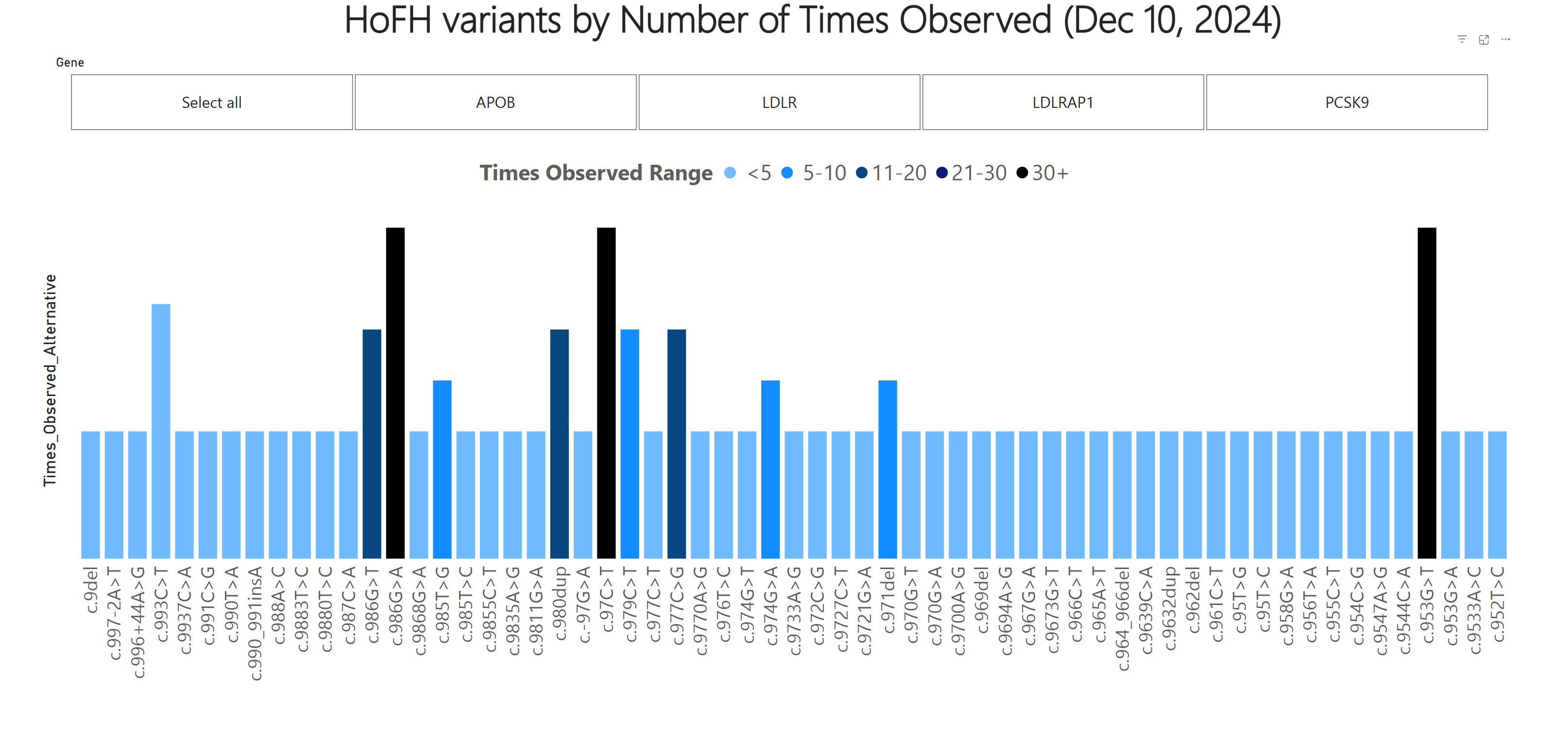
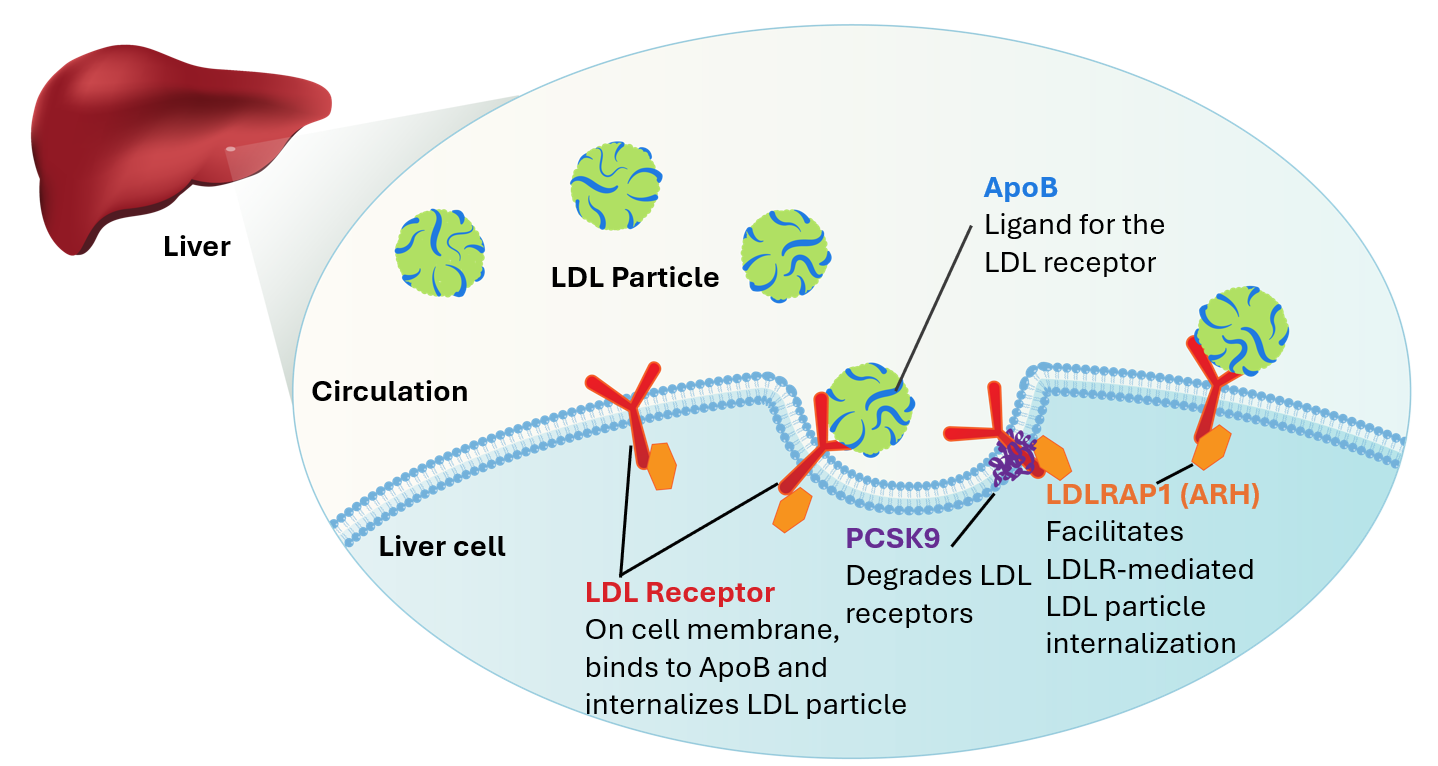
Homozygous familial hypercholesterolemia (HoFH) is a rare inherited hyperlipidemia leading to premature atherosclerotic disease and cardiac events due to extremely high circulating levels of low-density lipoprotein cholesterol (LDL-C) (1). HoFH is caused by bi-allelic pathogenic variants in familial hypercholesterolemia (FH)-causing genes, resulting in dangerously high levels (>400 mg/dL or >10 mmol/L) of LDL-C. The function of these genes is to control how the body regulates cholesterol levels in the blood (2).
Extremely high levels of cholesterol result in plaque deposits in arteries (atherosclerosis) and often leads to serious cardiovascular complications in childhood, like MI and stroke (3). Buildup of cholesterol can also cause aortic valve disease in the heart, xanthomas around joints and on tendons, corneal arcus in the eyes, and xanthelasmas around the eyelids (4).
Given the semi-dominant pattern of inheritance, LDLR, APOB, and PCSK9 are also associated with heterozygous familial hypercholesterolemia (HeFH) in individuals with monoallelic pathogenic variants. Heterozygotes characteristically have elevated LDL-C and are at risk of premature atherosclerotic disease and cardiac events. Typically, individuals with the heterozygous form of disease have less severely elevated LDL-C and risk of cardiovascular disease is lower than individuals impacted by biallelic disease. LDL-C levels alone may not always discriminate between patients with monallelic vs. biallelic disease, making genetic testing a helpful tool to differentiate these two disorders (5). This database reports on FH-causing variants, making it useful for characterizing both HeFH and HoFH genotypes.